The Wrong Suspect in the Cell’s Burn Chamber
For decades, scientists blamed the Krebs cycle and fat-burning machinery for making mitochondria leak toxic peroxide. But under moderate beta-oxidation, the usual suspects are exonerated. The real mystery is not why the cell is burning fuel—it’s what else inside the mitochondrion is making the dangerous exhaust.
The information in this article is for educational purposes only and is not intended as medical advice. Always consult a qualified healthcare professional for medical questions.
Summary
This article challenges the long-held belief that the Krebs cycle and fat-burning pathways are the primary sources of toxic hydrogen peroxide (H2O2) in mitochondria. A new study reveals that during moderate fat metabolism, these familiar "engines" are not the main culprits. Read this article to understand: - **The metabolic alibi:** Why the Krebs cycle and beta-oxidation are exonerated under moderate conditions. - **Beyond the powerhouse:** How mitochondria function as complex signaling hubs rather than simple furnaces. - **Future therapies:** Why pinpointing the exact source of oxidative stress is critical for treating aging and neurodegenerative diseases.
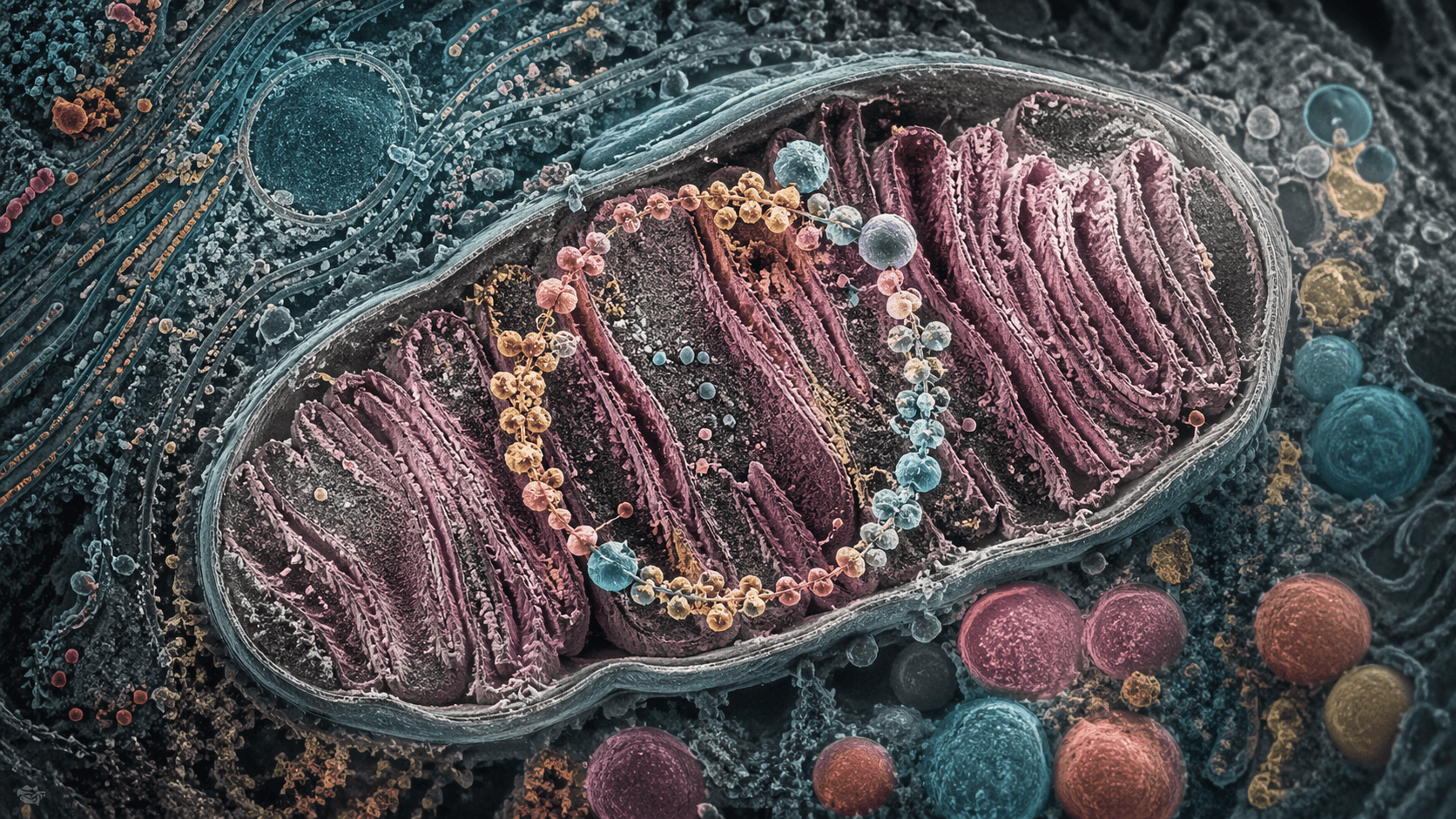
Mitochondria are often pictured as cellular powerhouses. They burn fuel, but reactive molecules can escape, potentially causing cellular stress. Suspicion often fell on the busiest parts of the machine — the Krebs cycle, which strips energy from carbon fuels, and beta-oxidation, which breaks down fats.
Now that familiar picture has a problem. Under moderate fat metabolism, the obvious suspects appear not to be the chief culprits. A study summarized by its title and findings reports that the Krebs cycle and mitochondrial fatty acid oxidation are not major producers of mitochondrial hydrogen peroxide under moderate beta-oxidation. Hydrogen peroxide, or H2O2, is one of the key reactive oxygen species that cells must control.
The finding does not absolve mitochondria. It does something more interesting: It offers an alibi for some of the most famous machinery inside them, under specific conditions. If the cell is burning fat at a moderate pace and peroxide still appears, then the mystery shifts. The question is no longer simply why fuel combustion leaks dangerous exhaust. It is what other mitochondrial process is producing it.
The engine that became a suspect
Mitochondria, central to cellular energy and function, are also implicated in various aspects of cellular health and disease. They consume oxygen. They process the carbon skeletons of sugars, fats and proteins. They generate much of the cell’s ATP, the small molecule that powers molecular work. In humans, this demand is staggering: The body turns over an amount of ATP roughly comparable to its own weight each day, and the brain, though only a small fraction of body mass, consumes a disproportionate share of that energy supply, according to a summary of mitochondrial energy metabolism and the Krebs cycle.
At the center of this metabolism sits the citric acid cycle, better known as the Krebs cycle. Hans Krebs described its logic in his 1953 Nobel lecture: A derivative of pyruvate joins with oxaloacetate to form citrate, beginning a repeating chemical loop that extracts energy from fuel and regenerates itself for another turn. Krebs emphasized that the cycle serves as a universal terminal pathway for carbohydrates, fats and proteins, a conserved engine that reveals nature’s economy of design in his account of the citric acid cycle’s discovery.
Given the cycle's central role in metabolism, it was often considered a key area of investigation when studying cellular stress. Fatty acids enter mitochondria and are dismantled through beta-oxidation. Those carbon fragments feed the Krebs cycle, which extracts energy from fuel. This process, involving oxygen and high-energy electrons, was often considered a potential site for the formation of reactive oxygen species, including hydrogen peroxide. This led to the assumption that increased fuel burning, particularly fat burning, would result in greater mitochondrial reactive oxygen species. The Krebs cycle and beta-oxidation were often considered prime suspects for reactive oxygen species production.
A metabolic alibi
The new result turns on a distinction that matters in biology but often disappears in shorthand: conditions. Cells do not run metabolism at one speed. They idle, sprint, fast, feast, divide, specialize and repair. The study highlights that a pathway's activity in producing reactive oxygen species can vary significantly with metabolic conditions.
The study at the heart of this story focuses on moderate beta-oxidation — a level of fat burning. Under those conditions, the researchers found that neither the Krebs cycle nor mitochondrial fatty acid oxidation was a major source of mitochondrial H2O2 during moderate levels of fat metabolism.
That statement may sound narrow. In fact, it challenges a common explanatory approach. When cells show signs of oxidative stress during fat metabolism, it is tempting to trace the damage backward to the fuel-burning pathway itself. Fat enters mitochondria; mitochondria generate peroxide; therefore fat oxidation must be the leak. But the reported finding breaks that chain of inference. It says that, at least under moderate beta-oxidation, the presence of fat metabolism does not identify beta-oxidation or the Krebs cycle as the main source of peroxide.
The result also highlights a distinction between two ideas. One is metabolic flux: the rate at which carbon and electrons move through pathways. The other is reactive oxygen species production: the side chemistry that creates oxidants. A pathway can carry substantial flux without being the dominant oxidant source. Conversely, other pathways or enzyme systems could be significant oxidant sources.

This is why the finding matters. It does not merely adjust a biochemical map. It changes where researchers should look when they try to understand damage.
The mitochondrion is not one machine
Part of the confusion comes from the word “mitochondria.” It suggests a single organelle with a single function: energy production. But mitochondria are crowded molecular cities. They host carbon metabolism, immune signaling, and communication with the nucleus through the signaling roles of TCA cycle metabolites.
Modern mitochondrial biology has pushed far beyond the old “powerhouse” metaphor. TCA cycle metabolites such as acetyl-CoA, alpha-ketoglutarate and succinate act as signals that can reshape gene expression, oxygen sensing, immune behavior and cancer biology, as described in a review of TCA metabolites as mitochondrial communication signals. These molecules are not passive intermediates. They help tell the cell what state it is in and what program it should run.
That signaling role complicates the search for oxidative stress. A metabolite can influence gene regulation without being the source of peroxide. A mitochondrion is less like a furnace than like a chemical city whose power plant, communications network and waste-control system share the same streets.
If beta-oxidation and the Krebs cycle are not the major H2O2 producers under moderate fat burning, attention turns to other mitochondrial machinery. Candidate sources could include other mitochondrial complexes or alternative pathways. The key point is not that one new culprit has already been convicted. It is that the old suspects no longer explain the case by default.
That distinction should force more careful experiments. Researchers may need to consider not only whether a cell is oxidizing fat, but also which mitochondrial redox states are active and which enzymes might be involved in oxygen reactions. Peroxide production is not a shadow automatically cast by metabolism. It is a biochemical event with a location, a mechanism and a context.
Why peroxide is not just poison
Hydrogen peroxide occupies an uneasy position in biology. It is dangerous enough that cells maintain systems to remove it.
This helps explain why identifying its source matters. If H2O2 were merely random chemical smoke, then reducing it everywhere might seem sensible. But if its production is regulated, understanding its source is crucial for developing targeted interventions.
The new finding therefore speaks to a broader shift in how metabolism is understood. The Krebs cycle is no longer only a grinder of fuel. Its intermediates can act as messengers that influence epigenetics and cell fate through mechanisms such as histone acetylation and oxygen-sensing pathways. Mitochondrial dysfunction is no longer a vague decline in energy. It may involve failures in signaling and redox balance.
If researchers attribute oxidative damage to the central carbon engine when another pathway is responsible, it risks misdirecting efforts to understand disease mechanisms and develop effective interventions.
Aging, disease and a misplaced blame
Mitochondrial oxidative stress has been a concept explored in aging research. The hypothesis that mitochondrial metabolism generates reactive oxygen species, which then damage the cell and contribute to aging, has been a subject of extensive investigation. The modern field is more nuanced. Aging is now described through interacting hallmarks that include genomic instability, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intercellular communication and other processes. A recent update expanded the framework to 12 hallmarks, adding compromised autophagy, microbiome dysbiosis and altered mechanical properties in an expanded account of the hallmarks of aging.
Mitochondrial dysfunction is recognized as one of these hallmarks, highlighting its interconnectedness with other aging mechanisms. They supply energy, regulate stress responses, influence inflammation and help determine whether damaged cells recover or die. In neurodegenerative disease, some researchers have argued that mitochondrial dysfunction and declining NAD+ levels may precede classic protein aggregates, shifting attention from plaques alone to the energetic state of vulnerable neurons in discussions of energy deficits in Alzheimer’s and Parkinson’s disease.
But if oxidative stress is part of the story, precision matters. “Mitochondrial ROS” is too broad a label. It can obscure the difference between a Krebs cycle enzyme, a fat-oxidation step, or other mitochondrial complexes and alternative pathways. Therapies based on an incomplete understanding may therefore lack precision.
The new result does not say that fat metabolism is harmless, that mitochondria do not generate peroxide, or that the Krebs cycle never contributes to oxidative stress. Biology rarely grants such sweeping verdicts. It says something more specific and more useful: Under moderate beta-oxidation, the canonical fuel-burning pathways are not the major producers of mitochondrial H2O2 according to the reported finding.
That specificity is the beginning of a better investigation. It narrows the search. It demands that mitochondrial oxidative stress be traced to mechanisms rather than assumed from metabolic activity. And it prompts a re-evaluation of long-held assumptions.
The cell’s burn chamber may still produce dangerous exhaust. But the furnace itself may not be the one leaking.
Also in Aging

New Facts About the Genetics of Longevity Uncovered
For decades, statistical noise masked the genetic drivers of human aging. Now, a mathematical correction has revealed that our intrinsic lifespan is highly heritable—and driven by reversible biological software.
